A.M.C. Davies
Norwich Near Infrared Consultancy, 10 Aspen Way, Cringleford, Norwich NR4 6UA, UK
Lorsque vous tendez la main vers un feu brûlant, vous « sentez » la chaleur émise par le feu mais que se passe-t-il ? Le feu émet de la lumière et des radiations infrarouges (IR) ; dans un feu, la plupart de ces radiations sont des radiations infrarouges proches (NIR). Une partie du rayonnement proche infrarouge est absorbée par les molécules d’eau de votre peau. Cela augmente la température de l’eau et entraîne une augmentation de la température des tissus environnants, qui est détectée par les nerfs de votre peau. Ce rayonnement a été découvert en 1800 par William Herschel, un musicien et astronome amateur très réputé (il a découvert la planète Uranus), car il voulait savoir si une couleur particulière était associée à la chaleur de la lumière du soleil. Il a constaté que le maximum de chaleur se situait au-delà de l’extrémité rouge du spectre. Herschel ne pouvait croire que la lumière et sa « chaleur rayonnante » étaient liées, mais il avait tort. En 1835, Ampère avait démontré que la seule différence entre la lumière et ce qu’il appelait le « rayonnement infrarouge » était leur longueur d’onde. Puis, en 1864, James Maxwell a écrit : « Cette vitesse est si proche de celle de la lumière qu’il semble que nous ayons de fortes raisons de conclure que la lumière elle-même (y compris la chaleur rayonnante et les autres rayonnements) est une perturbation électromagnétique sous forme d’ondes qui se propagent dans le champ électromagnétique selon les lois électromagnétiques ». Ce que nous appelons aujourd’hui le spectre électromagnétique est représenté sur la figure 1.

Histoire précoce de l’étude des absorptions infrarouges
Les premiers spectres infrarouges (proches) ont été mesurés en 1881 par Abney et Festing à l’aide de plaques photographiques. Non seulement ils ont produit les premiers spectres mais ils ont également suggéré, à juste titre, que les absorptions étaient liées à la composition chimique des liquides qu’ils étudiaient. Le pionnier le plus important de la spectroscopie IR est William W. Coblentz. En 1905, il a publié le résultat d’une vaste étude de composés dont il avait enregistré les spectres de 1000 nm à 16 000 nm. Les travaux de Coblentz ont constitué une percée dans la mesure où les chercheurs ont pu établir un lien entre le caractère des groupes d’atomes au sein des molécules et des absorptions spécifiques dans l’infrarouge moyen (2500-50 000 nm). Ces absorptions sont le résultat d’interactions avec les vibrations fondamentales des liaisons chimiques associées aux atomes des groupes. Nous pouvons considérer les liaisons chimiques comme des ressorts faibles qui maintiennent ensemble deux atomes ou plus. Ces ressorts vibrent naturellement et lorsqu’on ajoute de l’énergie au système, ils vibrent plus énergiquement. Cependant, les atomes des molécules sont limités par la mécanique quantique, de sorte que seuls quelques niveaux d’énergie spécifiques sont autorisés. Si nous n’avons que deux atomes, la seule vibration sera perçue comme un étirement. Lorsque trois atomes ou plus sont impliqués, les liaisons peuvent également se plier, ce qui donne lieu à toute une série de vibrations différentes. Les vibrations d’étirement nécessitent plus d’énergie que les vibrations de flexion, mais il y aura également des variations dans les besoins énergétiques des vibrations de flexion. Les différentes liaisons chimiques (comme O-H, C-H et N-H) varient en termes de force et donc de quantité d’énergie requise pour que la vibration de la liaison passe d’un niveau à l’autre. Cette variation d’énergie sera visible dans un spectre sous la forme d’une série d’absorptions à différentes longueurs d’onde. En regardant le spectre, nous pouvons déduire quelles sont les vibrations qui se produisent et donc travailler sur la structure de la molécule (ou des groupes d’atomes présents).
L’une des propriétés très utiles des spectres IR moyen est que la région allant de 8500 nm à 12 500 nm est très caractéristique de la molécule mesurée et cette région est connue comme la région des « empreintes digitales » car elle peut être utilisée pour confirmer l’identité de nombreuses substances pures. Si l’étude de la spectroscopie dans l’infrarouge moyen a continué à se développer, surtout après la Seconde Guerre mondiale, l’intérêt pour le proche infrarouge s’est limité à des mesures quantitatives de l’eau, de quelques composés organiques simples et à quelques rares études de protéines spécifiques. Personne ne le considérait utile pour caractériser les échantillons et il était considéré comme trop complexe pour être utilisé dans l’analyse quantitative.
Absorptions dans la région NIR
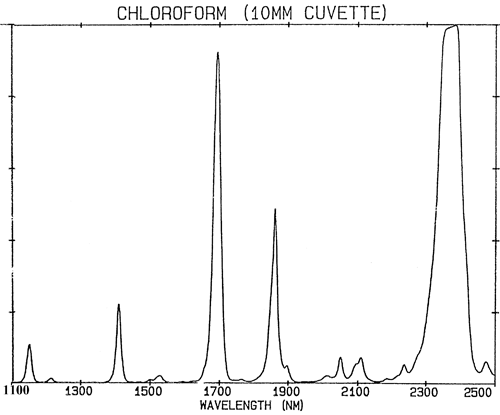
Si les liaisons chimiques se comportaient exactement comme des ressorts faibles, alors la mécanique quantique limiterait leur vibration à seulement deux états et il y aurait très peu d’absorptions dans la région NIR. Les absorptions dans la région NIR (780-2500 nm) sont générées à partir des vibrations fondamentales par deux processus : les harmoniques et les combinaisons. Les harmoniques peuvent être considérées comme des harmoniques. Ainsi, chaque fondamentale produira une série d’absorptions à des multiples (approximativement entiers) de la fréquence (la fréquence est l’inverse de la longueur d’onde). Les combinaisons sont plus complexes. Les absorptions dans le proche infrarouge sont à un niveau d’excitation plus élevé et nécessitent donc plus d’énergie qu’une absorption fondamentale. Les combinaisons résultent du partage de l’énergie NIR entre deux ou plusieurs absorptions fondamentales. Bien que le nombre d’harmoniques possibles d’un groupe d’absorptions fondamentales dans une molécule soit limité à quelques-unes, un très grand nombre de combinaisons sera observé. L’effet combiné de toutes ces absorptions fait que de nombreux spectres NIR sont plutôt inintéressants et ne comportent que quelques pics assez larges. La figure 2 est un spectre NIR du chloroforme, CHCl3, la molécule ne contient qu’un seul atome d’hydrogène mais toutes les absorptions de son spectre sont causées par cet unique atome.
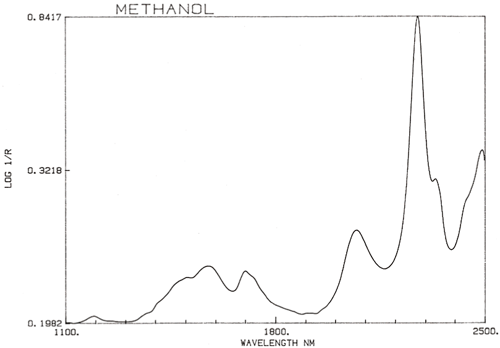
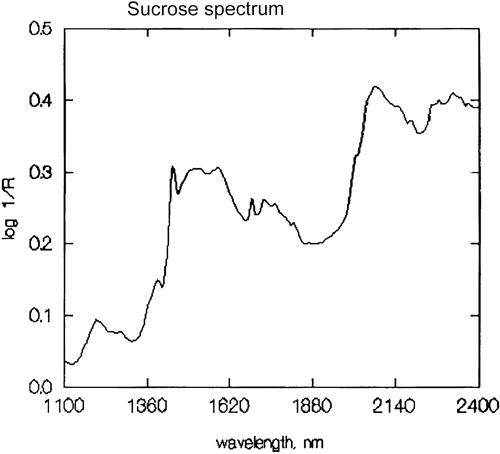
C’est une généralisation importante que la spectroscopie NIR est dominée par l’hydrogène. La figure 3 est un spectre de méthanol, CH3OH, qui contient quatre atomes d’hydrogène (mais trois sont équivalents) et ce spectre ressemble beaucoup plus à un spectre NIR typique avec des pics larges. La figure 4 est un spectre de saccharose, C12H24O12, qui montre des zones d’absorption très larges mais aussi des pics assez étroits. Il est important de réaliser que toutes ces absorptions larges sont causées par de multiples absorptions étroites qui se chevauchent. Les spectres NIR sont beaucoup plus complexes qu’il n’y paraît.
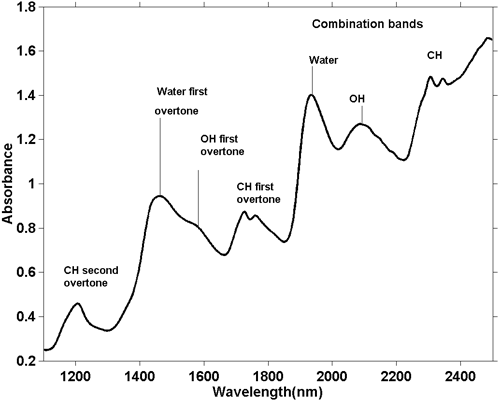
Bien que les spectres NIR soient plus compliqués, il est possible de faire quelques observations générales. Comme une absorption fondamentale d’étirement O-H est différente d’une absorption fondamentale d’étirement C-H, la série d’harmoniques générées par ces absorptions sera également différente. Il en va de même pour les bandes combinées. Les bandes combinées les plus courantes (et les plus énergétiques) proviennent de combinaisons d’étirement et de flexion dans le même groupe. Ainsi, nous voyons des absorptions dues à la combinaison d’un étirement O-H avec un coude O-H et d’un étirement C-H avec un coude C-H, et ces absorptions apparaissent à des positions différentes dans le spectre. La figure 5 représente le spectre NIR d’un échantillon de pâte à biscuits. La pâte à biscuits contient plusieurs ingrédients, chacun d’entre eux contenant de nombreuses molécules différentes. Ce spectre contient donc des centaines, voire des milliers d’absorptions, mais nous voyons l’intégration de toutes ces absorptions et il semble qu’il n’y en ait que quelques-unes. A partir de leur position, nous pouvons dire en termes généraux la cause de l’absorption, comme indiqué sur la figure.
Lorsque la complexité de l’absorption NIR a été réalisée pour la première fois et comparée aux spectres mid-IR relativement plus faciles à comprendre, la plupart des chercheurs ont pensé qu’il y avait peu à gagner en étudiant la spectroscopie NIR. La région a été négligée et les étudiants ont été informés à tort qu’il n’y avait rien à gagner à étudier la région NIR. De nombreux étudiants sont encore convaincus de la même chose. Les conditions requises étaient les suivantes : des spectromètres à très faible bruit, un ordinateur électronique, l’application de techniques mathématiques (chimiométrie) et un génie pour rassembler le tout. Cet homme s’appelait Karl Norris, un ingénieur travaillant pour l’USDA à Beltsville. Comme on ne lui avait pas enseigné la spectroscopie, il ne savait pas qu’il n’y avait rien à gagner dans la région du proche infrarouge. Ainsi, à l’instar de Herschel qui cherchait quelque chose là où il n’y avait rien, Norris a mis au point des instruments et utilisé des ordinateurs pour démontrer que la région du proche infrarouge était très utile pour l’analyse quantitative, en particulier des échantillons agricoles. L’une des raisons pour lesquelles l’analyse dans le proche infrarouge est si utile est qu’elle peut utiliser l’énergie réfléchie, ce qui signifie que l’analyse dans le proche infrarouge peut être effectuée avec peu ou pas de préparation de l’échantillon. L’énergie réfléchie est complexe. D’abord, parce qu’elle comporte deux composantes, spéculaire (ou miroir) et diffuse. Dans le contexte de la spectroscopie NIR, la composante spéculaire ne donne aucune information. La composante diffuse dépend de la nature physique de l’échantillon, la taille des particules étant particulièrement importante. La variation des paramètres physiques d’un échantillon entraîne des changements dans le spectre, de sorte que le spectre observé est un mélange d’informations chimiques et physiques.
L’utilisation de l’énergie réfléchie a été imposée à Karl Norris. Si elle rend possible l’analyse NIR d’une très large gamme d’échantillons, elle ajoute une autre couche de complication. Une théorie mathématique complète de la spectroscopie de réflexion n’est pas encore disponible, mais une bonne pratique expérimentale et l’utilisation de techniques mathématiques ont permis d’utiliser la spectroscopie de réflexion NIR en chimie analytique. Comme la technique peut être appliquée avec peu ou pas de préparation d’échantillon, les temps d’analyse sont réduits de plusieurs heures à quelques minutes et, de plus, plusieurs résultats analytiques peuvent être obtenus à partir des mêmes données NIR alors que l’analyse conventionnelle nécessiterait souvent une autre technique et plus d’heures de travail. Il est cependant nécessaire de développer des calibrations qui nécessitent de nombreux échantillons, de nombreuses heures de travail et des milliers (ou probablement des millions) de calculs informatiques. Avec ce genre d’attributs, il n’est pas surprenant que 40 ans après les recherches révolutionnaires, une très large gamme d’analyses puisse être réalisée par la spectroscopie NIR.
Ce qui est surprenant, c’est que malgré le succès de l’analyse spectroscopique NIR, dans le monde entier, il y a très peu de départements universitaires de chimie qui ont un programme de recherche en spectroscopie NIR. Par conséquent, la majorité des étudiants en chimie quittent l’université sans aucune connaissance du NIR, à l’exception peut-être de l’opinion désuète selon laquelle il n’y a rien d’utile à apprendre sur la région du NIR.
.