A.M.C. Davies
Norwich Near Infrared Consultancy, 10 Aspen Way, Cringleford, Norwich NR4 6UA, UK
p>Quando se estende a mão a um fogo ardente, “sente-se” o calor emitido pelo fogo, mas o que está a acontecer? O fogo emite luz e radiação infravermelha (IR); de um incêndio a maior parte desta é radiação quase infravermelha (NIR). Parte da radiação NIR é absorvida por moléculas de água na sua pele. Isto aumenta a temperatura da água e resulta num aumento da temperatura do tecido circundante que é detectado pelos nervos na sua pele. Esta radiação foi descoberta em 1800 por William Herschel, músico e astrónomo amador muito bem sucedido (descobriu o planeta Urano) porque queria saber se alguma cor em particular estava associada ao calor da luz solar. Descobriu que o calor máximo estava para além da extremidade vermelha do espectro. Herschel não podia acreditar que a luz e o seu “calor radiante” estivessem relacionados, mas estava errado. Em 1835 Ampere tinha demonstrado que a única diferença entre a luz e o que ele denominou “radiação infravermelha” era o seu comprimento de onda. Então em 1864 James Maxwell escreveu “Esta velocidade é tão próxima da da luz que parece que temos fortes razões para concluir que a própria luz (incluindo calor radiante e outras radiações) é uma perturbação electromagnética sob a forma de ondas propagadas através do campo electromagnético de acordo com as leis electromagnéticas”. O que agora chamamos espectro electromagnético é mostrado na Figura 1.

A história do estudo das absorções de infravermelhos
Os primeiros espectros de infravermelhos (próximos) foram medidos em 1881 por Abney e Festing usando placas fotográficas. Não só produziram os primeiros espectros como também sugeriram, correctamente, que as absorções estavam relacionadas com a composição química dos líquidos que investigaram. O pioneiro mais importante da espectroscopia IR foi William W. Coblentz. Em 1905 publicou o resultado de um grande estudo de compostos cujos espectros tinha registado de 1000 nm a 16.000 nm. O trabalho de Coblentz foi um avanço na medida em que os investigadores foram capazes de relacionar o carácter de grupos de átomos dentro das moléculas como estando relacionados com absorções específicas no meio do IR (2500-50.000 nm). Estas absorções são o resultado de interacções com as vibrações fundamentais das ligações químicas associadas com os átomos dos grupos. Podemos pensar em ligações químicas como molas fracas que mantêm juntos dois ou mais átomos, estas molas vibrarão naturalmente e quando a energia é adicionada ao sistema, então vibrarão mais energeticamente. Contudo, os átomos nas moléculas são limitados pela mecânica quântica, de modo que apenas alguns níveis específicos de energia são permitidos. Se tivermos apenas dois átomos, então a única vibração será vista como um alongamento. Quando três ou mais átomos estão envolvidos, as ligações também se podem dobrar, dando origem a toda uma série de vibrações diferentes. As vibrações de alongamento requerem mais energia do que as vibrações de flexão, mas também haverá variação nas necessidades energéticas das vibrações de flexão. Diferentes ligações químicas (como O-H, C-H e N-H) variam em força e, portanto, a quantidade de energia necessária para que a vibração da ligação se mova de um nível para o outro. Esta variação na energia será vista num espectro como uma série de absorções em diferentes comprimentos de onda. Olhando para o espectro, podemos deduzir quais as vibrações que estão a ocorrer e, portanto, calcular a estrutura da molécula (ou grupos de átomos presentes).
Uma das propriedades muito úteis dos espectros de meio-IR é que a região de 8500 nm a 12.500 nm é muito característica para a molécula medida e esta região é conhecida como a região da “impressão digital” porque pode ser utilizada para confirmar a identidade de muitas substâncias puras. Enquanto que o estudo da espectroscopia de mid-IR continuou a crescer, especialmente após a Segunda Guerra Mundial, o interesse no NIR estendeu-se às medições quantitativas da água, a alguns compostos orgânicos simples e a muito poucos estudos de proteínas específicas. Ninguém o considerou útil para caracterizar amostras e foi considerado demasiado complexo para ser utilizado em análises quantitativas.
Absorções na região NIR
Se as ligações químicas se comportassem exactamente como molas fracas, a mecânica quântica restringiria a sua vibração a apenas dois estados e haveria muito poucas absorções na região NIR. As absorções na região NIR (780-2500 nm) são geradas a partir de vibrações fundamentais por dois processos; sobre-tons e combinações. Os excessos podem ser pensados como harmónicos. Assim, cada fundamental produzirá uma série de absorções em múltiplos (aproximadamente inteiros) da frequência (a frequência é o recíproco de comprimento de onda). As combinações são bastante mais complexas. As absorções NIR estão num estado de maior excitação, pelo que requerem mais energia do que uma absorção fundamental. As combinações resultam da partilha da energia NIR entre duas ou mais absorções fundamentais. Embora o número de possíveis sobretons de um grupo de absorções fundamentais numa molécula seja limitado a poucas, será observado um número muito grande de combinações. O efeito de todas estas absorções combina para que muitos espectros de NIR pareçam bastante desinteressantes e consistam apenas em poucos picos bastante amplos. A figura 2 é um espectro NIR de clorofórmio, CHCl3, a molécula contém apenas um átomo de hidrogénio mas toda a absorção no seu espectro é causada por este único átomo.
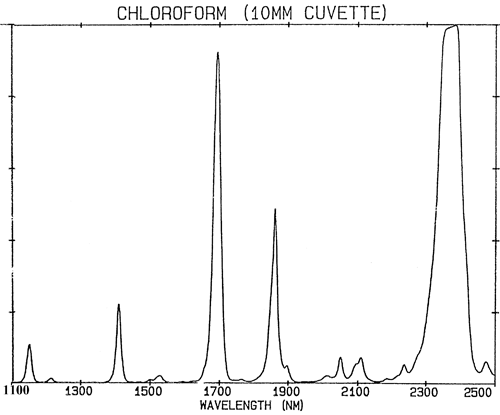
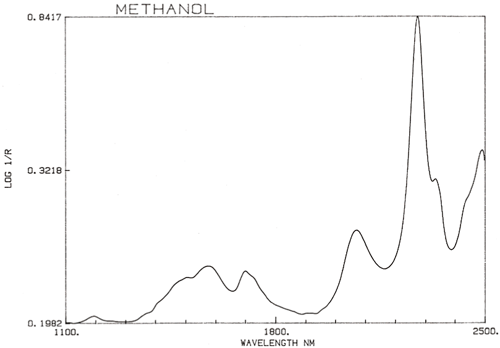
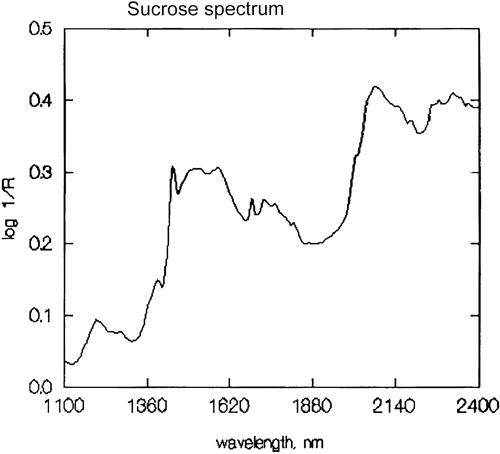
É uma generalização importante que a espectroscopia NIR é dominada pelo hidrogénio. A figura 3 é um espectro de metanol, CH3OH, que contém quatro átomos de hidrogénio (mas três são equivalentes) e este espectro é muito mais parecido com um espectro típico do NIR com picos amplos. A figura 4 é um espectro de sacarose, C12H24O12, que mostra áreas de absorção muito amplas mas também alguns picos bastante estreitos. É importante perceber que todas estas amplas absorções são causadas por múltiplas absorções estreitas e sobrelaçadas. Os espectros NIR são muito mais complexos do que parecem.
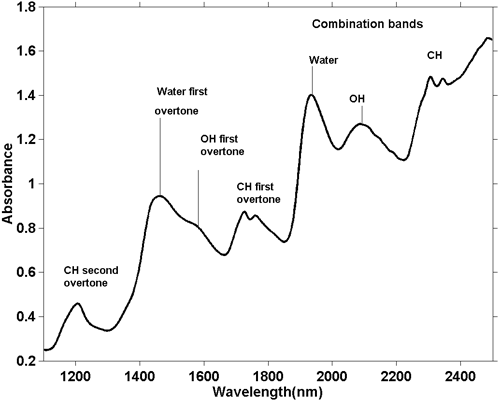
P>Embora os espectros NIR sejam mais complicados, é possível fazer algumas observações gerais. Como uma absorção de alongamento O-H fundamental é diferente de um alongamento C-H fundamental, então a série de sobretons gerados por estas absorções também será diferente. O mesmo se aplica às bandas combinadas. As bandas de combinação mais comuns (e energéticas) surgem a partir de combinações de estiramento e dobra no mesmo grupo. Assim, vemos absorções devidas à combinação de estiramento O-H com curva O-H e estiramento C-H com curva C-H e estas ocorrem em diferentes posições no espectro. A figura 5 é o espectro NIR de uma amostra de massa de biscoito. A massa de biscoito contém vários ingredientes, cada um dos quais contém muitas moléculas diferentes, pelo que este espectro contém centenas se não milhares de absorções, mas vemos a integração de todas elas e parece haver apenas algumas absorções amplas. Da sua posição podemos dizer em termos gerais a causa da absorção, como indicado na figura.
Quando a complexidade da absorção do NIR foi realizada pela primeira vez e comparada com os espectros relativamente mais fáceis de compreender do meio do AR, pensou-se pela maioria dos investigadores que havia pouco a ganhar com o estudo da espectroscopia do NIR. A região tornou-se negligenciada e os estudantes foram erradamente instruídos de que não havia nada a ganhar com o estudo da região do NIR. Muitos estudantes ainda estão a ser ensinados a mesma opinião. Os requisitos eram: espectrómetros de ruído muito baixo, o computador electrónico, a aplicação de técnicas matemáticas (quimiometria) e um génio para reunir tudo isto. O homem era Karl Norris; um engenheiro que trabalhava para a USDA em Beltsville. Não lhe tinham ensinado espectroscopia, pelo que não sabia que não havia nada a ganhar na região do NIR. Assim, um pouco como Herschel que procurava algo onde não havia nada, Norris desenvolveu os instrumentos e utilizou computadores para demonstrar que a região do NIR era muito útil para análises quantitativas, particularmente de amostras agrícolas. Uma das razões pelas quais a análise NIR é tão útil é que pode utilizar energia reflectida e isto significa que a análise NIR pode ser feita com pouca ou nenhuma preparação de amostras. A energia reflectida é complexa. Em primeiro lugar, porque existem dois componentes, especular (ou tipo espelho) e difuso. No contexto da espectroscopia NIR, o componente especular não dá qualquer informação. O componente difuso depende da natureza física da amostra; o tamanho da partícula é particularmente importante. A variação dos parâmetros físicos de uma amostra provoca alterações no espectro de modo que o espectro observado é uma mistura de informação química e física.
A utilização de energia reflectida foi forçada a Karl Norris. Embora torne possível a análise NIR de uma gama muito mais vasta de amostras, acrescenta outra camada de complicação. Uma teoria matemática completa da espectroscopia de reflexão ainda não está disponível, mas foi considerada possível pela boa prática experimental e pela utilização de técnicas matemáticas para utilizar a espectroscopia de reflexão NIR para a química analítica. Como a técnica pode ser aplicada com pouca ou nenhuma preparação de amostras, os tempos de análise são reduzidos de horas para minutos e, além disso, vários resultados analíticos podem ser obtidos a partir dos mesmos dados NIR, enquanto a análise convencional requereria frequentemente outra técnica e mais horas de trabalho. No entanto, é necessário desenvolver calibrações que requerem muitas amostras, muitas horas de trabalho e milhares (ou provavelmente milhões) de cálculos informáticos. Com este tipo de atributos não é surpreendente que 40 anos após a investigação pioneira, uma gama muito ampla de análises possa ser alcançada pela espectroscopia NIR.
O que é surpreendente é que, apesar do sucesso da análise espectroscópica NIR, em todo o mundo há muito poucos departamentos de química universitária que tenham qualquer programa de investigação na espectroscopia NIR. Consequentemente, a maioria dos estudantes de química deixam a universidade sem qualquer conhecimento do NIR, com a possível excepção da visão antiquada de que não há nada útil para aprender sobre a região NIR.