A.M.C. Davies
Norwich Near Infrared Consultancy, 10 Aspen Way, Cringleford, Norwich NR4 6UA, UK
Cuando se acerca la mano a un fuego ardiente se «siente» el calor que emite el fuego, pero ¿qué ocurre? El fuego emite luz y radiación infrarroja (IR); de un fuego la mayor parte es radiación infrarroja cercana (NIR). Parte de la radiación NIR es absorbida por las moléculas de agua de la piel. Esto eleva la temperatura del agua y provoca un aumento de la temperatura en el tejido circundante que es detectado por los nervios de la piel. Esta radiación fue descubierta en 1800 por William Herschel, músico y astrónomo aficionado de gran éxito (descubrió el planeta Urano), porque quería saber si algún color concreto estaba asociado al calor de la luz solar. Descubrió que el máximo de calor estaba más allá del extremo rojo del espectro. Herschel no podía creer que la luz y su «calor radiante» estuvieran relacionados, pero se equivocaba. En 1835, Ampere había demostrado que la única diferencia entre la luz y lo que él denominó «radiación infrarroja» era su longitud de onda. Luego, en 1864, James Maxwell escribió: «Esta velocidad es tan parecida a la de la luz que parece que tenemos razones de peso para concluir que la luz misma (incluyendo el calor radiante y otras radiaciones) es una perturbación electromagnética en forma de ondas que se propagan a través del campo electromagnético según las leyes electromagnéticas». Lo que ahora llamamos espectro electromagnético se muestra en la figura 1.

Historia temprana del estudio de las absorciones infrarrojas
Los primeros espectros infrarrojos (cercanos) fueron medidos en 1881 por Abney y Festing utilizando placas fotográficas. No sólo produjeron los primeros espectros sino que también sugirieron, correctamente, que las absorciones estaban relacionadas con la composición química de los líquidos que investigaban. El pionero más importante de la espectroscopia IR fue William W. Coblentz. En 1905 publicó el resultado de un amplio estudio de compuestos cuyos espectros había registrado desde 1000 nm hasta 16.000 nm. El trabajo de Coblentz supuso un gran avance, ya que los investigadores pudieron relacionar el carácter de los grupos de átomos dentro de las moléculas con absorciones específicas en el infrarrojo medio (2500-50.000 nm). Estas absorciones son el resultado de las interacciones con las vibraciones fundamentales de los enlaces químicos asociados a los átomos de los grupos. Podemos pensar en los enlaces químicos como resortes débiles que mantienen unidos a dos o más átomos, estos resortes vibrarán de forma natural y cuando se añada energía al sistema entonces vibrarán más energéticamente. Sin embargo, los átomos de las moléculas están limitados por la mecánica cuántica, de modo que sólo se permiten unos pocos niveles de energía específicos. Si sólo hay dos átomos, la única vibración se verá como un estiramiento. Cuando hay tres o más átomos, los enlaces también pueden doblarse, dando lugar a toda una serie de vibraciones diferentes. Las vibraciones de estiramiento requieren más energía que las de flexión, pero también habrá variaciones en los requisitos energéticos de las vibraciones de flexión. Los diferentes enlaces químicos (como el O-H, el C-H y el N-H) varían en fuerza y, por tanto, en la cantidad de energía necesaria para que la vibración del enlace pase de un nivel al siguiente. Esta variación en la energía se verá en un espectro como una serie de absorciones a diferentes longitudes de onda. Observando el espectro podemos deducir qué vibraciones se están produciendo y, por lo tanto, averiguar la estructura de la molécula (o de los grupos de átomos presentes).
Una de las propiedades muy útiles de los espectros del infrarrojo medio es que la región de 8500 nm a 12.500 nm es muy característica para la molécula medida y esta región se conoce como la región de la «huella dactilar» porque puede utilizarse para confirmar la identidad de muchas sustancias puras. Aunque el estudio de la espectroscopia del IR medio siguió creciendo, especialmente después de la Segunda Guerra Mundial, el interés por el IRN se limitó a las mediciones cuantitativas del agua, unos pocos compuestos orgánicos simples y unos pocos estudios de proteínas específicas. Nadie la consideraba útil para caracterizar muestras y se consideraba demasiado compleja para su uso en análisis cuantitativos.
Absorciones en la región NIR
Si los enlaces químicos se comportaran exactamente como resortes débiles, entonces la mecánica cuántica restringiría su vibración a sólo dos estados y habría muy pocas absorciones en la región NIR. Las absorciones en la región NIR (780-2500 nm) se generan a partir de las vibraciones fundamentales mediante dos procesos: los sobretonos y las combinaciones. Los sobretonos pueden considerarse como armónicos. Así, cada fundamental producirá una serie de absorciones en múltiplos (aproximadamente enteros) de la frecuencia (la frecuencia es el recíproco de la longitud de onda). Las combinaciones son bastante más complejas. Las absorciones NIR se encuentran en un estado de excitación superior, por lo que requieren más energía que una absorción fundamental. Las combinaciones surgen al compartir la energía NIR entre dos o más absorciones fundamentales. Aunque el número de posibles sobretonos de un grupo de absorciones fundamentales en una molécula está limitado a unos pocos, se observará un número muy grande de combinaciones. El efecto de todas estas absorciones se combina para hacer que muchos espectros NIR parezcan poco interesantes y estén formados sólo por unos pocos picos bastante amplios. La figura 2 es un espectro NIR del cloroformo, CHCl3, la molécula contiene sólo un átomo de hidrógeno pero toda la absorción en su espectro es causada por este único átomo.
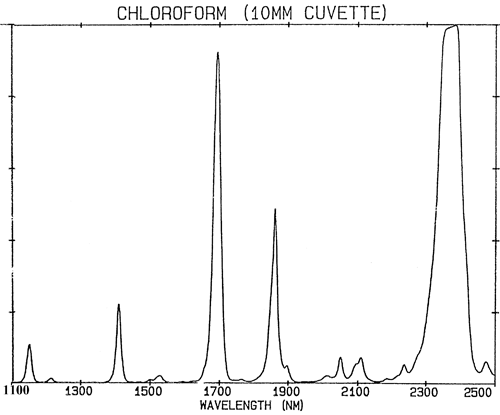
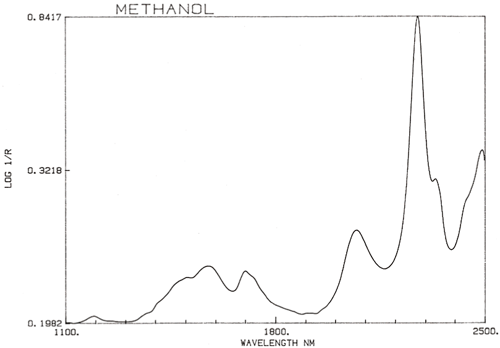
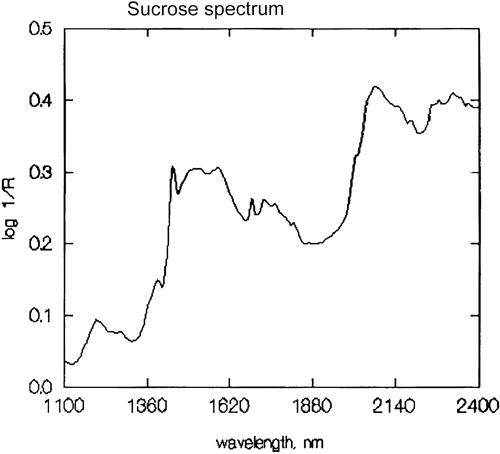
Es una generalización importante que la espectroscopia NIR esté dominada por el hidrógeno. La figura 3 es un espectro de metanol, CH3OH, que contiene cuatro átomos de hidrógeno (pero tres son equivalentes) y este espectro es mucho más parecido a un espectro NIR típico con picos amplios. La figura 4 es un espectro de sacarosa, C12H24O12, que muestra áreas de absorción muy amplias pero también algunos picos bastante estrechos. Es importante tener en cuenta que todas estas absorciones amplias están causadas por múltiples absorciones estrechas y superpuestas. Los espectros NIR son mucho más complejos de lo que parecen.
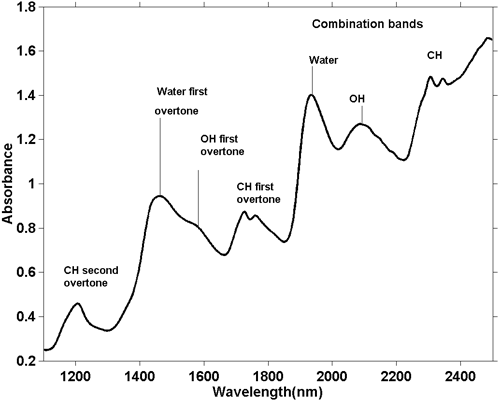
Aunque los espectros NIR son más complicados, es posible hacer algunas observaciones generales. Como una absorción de estiramiento O-H fundamental es diferente a un estiramiento C-H fundamental, entonces la serie de sobretonos generados por estas absorciones también será diferente. Lo mismo ocurre con las bandas combinadas. Las bandas combinadas más comunes (y energéticas) surgen de las combinaciones de estiramiento y flexión en el mismo grupo. Así, vemos las absorciones debidas a la combinación del estiramiento O-H con la flexión O-H y el estiramiento C-H con la flexión C-H y éstas se producen en diferentes posiciones en el espectro. La figura 5 es el espectro NIR de una muestra de masa de galleta. La masa de galletas contiene varios ingredientes, cada uno de los cuales contiene muchas moléculas diferentes, por lo que este espectro contiene cientos, si no miles, de absorciones, pero vemos la integración de todas ellas y parece que sólo hay unas pocas absorciones amplias. A partir de su posición podemos decir en términos generales la causa de la absorción, como se indica en la figura.
Cuando se comprendió por primera vez la complejidad de la absorción NIR y se comparó con los espectros relativamente más fáciles de entender del infrarrojo medio, la mayoría de los investigadores pensaron que había poco que ganar estudiando la espectroscopia NIR. La región se descuidó y se instruyó erróneamente a los estudiantes de que no había nada que ganar estudiando la región NIR. A muchos estudiantes se les sigue enseñando la misma opinión. Los requisitos eran: espectrómetros de muy bajo ruido, el ordenador electrónico, la aplicación de técnicas matemáticas (quimiometría) y un genio que lo reuniera todo. El hombre era Karl Norris; un ingeniero que trabajaba para el USDA en Beltsville. No le habían enseñado espectroscopia, por lo que no sabía que no había nada que ganar en la región NIR. Así que, al igual que Herschel, que buscaba algo donde no había nada, Norris desarrolló los instrumentos y utilizó los ordenadores para demostrar que la región NIR era muy útil para el análisis cuantitativo, sobre todo de muestras agrícolas. Una de las razones por las que el análisis NIR es tan útil es que puede utilizar la energía reflejada y esto significa que el análisis NIR puede realizarse con poca o ninguna preparación de la muestra. La energía reflejada es compleja. En primer lugar, porque hay dos componentes, el especular (o de espejo) y el difuso. En el contexto de la espectroscopia NIR, el componente especular no da ninguna información. El componente difuso depende de la naturaleza física de la muestra, siendo el tamaño de las partículas especialmente importante. La variación de los parámetros físicos de una muestra provoca cambios en el espectro, de modo que el espectro observado es una mezcla de información química y física.
El uso de la energía reflejada fue forzado por Karl Norris. Aunque hace posible el análisis NIR de una gama mucho más amplia de muestras, añade otra capa de complicación. Todavía no se dispone de una teoría matemática completa de la espectroscopia de reflexión, pero se ha descubierto que es posible utilizar la espectroscopia de reflexión NIR para la química analítica gracias a una buena práctica experimental y a la utilización de técnicas matemáticas. Como la técnica puede aplicarse con poca o ninguna preparación de la muestra, los tiempos de análisis se reducen de horas a minutos y, además, pueden obtenerse varios resultados analíticos a partir de los mismos datos NIR, mientras que el análisis convencional requeriría a menudo otra técnica y más horas de trabajo. Sin embargo, es necesario desarrollar calibraciones que requieren muchas muestras, muchas horas de trabajo y miles (o probablemente millones) de cálculos informáticos. Con este tipo de atributos no es de extrañar que 40 años después de la investigación pionera, se pueda lograr una gama muy amplia de análisis mediante la espectroscopia NIR.
Lo que sí es sorprendente es que, a pesar del éxito del análisis espectroscópico NIR, en todo el mundo hay muy pocos departamentos universitarios de química que tengan algún programa de investigación en espectroscopia NIR. En consecuencia, la mayoría de los estudiantes de química salen de la universidad sin ningún conocimiento sobre el NIR, con la posible excepción de la opinión anticuada de que no hay nada útil que aprender sobre la región NIR.